
 English
English 中文
中文
Behind the Paper--PanSyn: Elucidation of Pan-evolutionary and Regulatory Genome Architecture
Unraveling the structure and function of genomes lies at the core of decoding the history of life on Earth and biodiversity. The genome data generated by the Earth BioGenome Project has opened up a new era of comparative genomics, for which genome synteny analysis provides an important framework. Profiling genome synteny represents an essential step in elucidating genome architecture, regulatory blocks/elements, and their evolutionary history. Over the past years, comparative genome synteny analyses of both traditional model organisms and recently emerging non-model organisms have led to numerous major discoveries and breakthroughs1-6.
In the summer of 2019, I joined Prof. Wang's lab (http://www.molevolab.com) at Ocean University of China. Driven by the power of a new era in genomics, our team has carried out extensive research in comparative genomics. In 2017, our team incisively discovered that Yesso scallop has a slow-evolving or fossil-like genome using the developed chromosome-based macrosynteny pipeline3 (Behind the Paper: The “fossil” genome of scallop Patinopecten yessoensis). Then, we have made many important findings through genome synteny-based evolutionary analyses, including the origin and evolution of molluscan red-bloodedness7, the adaptive bird-like genome miniaturization8, and the ancient homomorphy of molluscan sex chromosomes9. At the same time, the genome macrosynteny has been customized as the database module specifically for molluscs (MolluscDB)10 and animal EvoDevo research (EDomics)11, which has been widely recognized and used by international scientific community. Notably, we discovered that while there were several well-known software tools for synteny analysis12-14, most were designed for profiling genome microsynteny at subchromosomal scales, requiring conserved gene orders and limiting comparisons to phylogenetically closely related species. However, understanding the macroevolution of genome architecture from ancestors to extant species depends on deep phylogenetic comparisons, involving the investigation of ancestral conserved linkages between orthologous genes independent of gene order2-4. Unfortunately, user-friendly and customizable bioinformatic tools for genome macrosynteny at chromosomal scales are largely lacking despite the usefulness of such approaches in inferring the ancestral karyotype evolution history in the animal kingdom2-4,11,15. The relevance of these tools is expected to increase in the future, especially with the advent of many more chromosome-scale genomes. It can thus be seen, development of new software tools for genome synteny is an inspiring trend. Therefore, our team's strong interest and extensive research experience in genome synteny led us to embark on the development of the software package for the deep profiling of genome synteny.
Against this background, my initial idea was to develop a software capable of efficiently performing macrosynteny analysis. However, just when I was about to complete the development of the macrosynteny analytical pipeline and thought that the project was nearing its end, a turning point in this project emerged during my daily discussions with Prof. Wang. He suggested that I could consider developing a new tool for comprehensive genome synteny, rather than just focusing on macrosynteny. At first, I couldn't help but wonder if this was a good idea. So, the question we need to think in this upgraded project is: Is it truly necessary and important to develop a new software for comprehensive synteny analysis? To answer the question, we reviewed the history of our understanding of genome synteny and researched fifty synteny-related tools. We found that many microsynteny software led to simple or limited function, unable to provide multi-faceted analysis. And most current approaches investigate genome synteny solely at either micro- or macro-evolution scales, and integrative analysis of both scales remains scarce7,8,16,17, preventing a comprehensive understanding of pan-evolutionary history. These findings suggest that that existing synteny tools were unable to keep up with the advancements and need in synteny research. Under this situation, developing a new tool exclusively for macrosynteny analysis is insufficient to address the deficiencies of current synteny analytical methods. These findings inspired us to move forward with the development of a new pipeline named PanSyn, designed for comprehensive synteny analysis by profiling both genome microsynteny and macrosynteny. Thus, I continued on the journey to develop and refine PanSyn.
With the multi-dimensional development of synteny analysis, it’s challenging for us to develop the most comprehensive and up-to-date genome synteny pipeline. Everything's hard in the beginning. A lot of effort was expended in the initial exploration and determination of the framework. Fortunately, our teams have extensive research experience as well as pipeline development to help me. One of the important turning points resulted from our team research on the scallop sex chromosomes. Our team unexpectedly found that combining functional genomic data with genome synteny helped to reveal a translocatable enhancer-based mechanism of sex chromosome turnover9 (Behind the Paper: The ‘forever-young’ secret of scallop sex chromosomes: exceptions to the ‘general’ rules). This suggests that it might be a good point for us to incorporate functional genomics datasets into our pipeline. And then through further literature research, we further confirm that integrating multi-dimensional functional omics data with genome synteny is crucial to understand the functional significance of genomic synteny, which also enable the efficient utilization of the massive increasing omics datasets from ENCODE and 3D/4D genome projects. These functional integrated approaches promise to reveal mechanisms of gene function and regulation, and to enable exploration of how synteny genes work together to modulate complex phenotypes18. Based on this idea, through my ongoing discussions with Prof. Wang and Prof. Li, we outlined the overall software framework into three parts: the comprehensive microsynteny analysis, the advanced macrosynteny analysis, and the functional integration of microsynteny and macrosynteny. Based on this framework, with relentless efforts, we dedicated approximately two years to complete the development of PanSyn (Fig. 1). It not only includes a comprehensive microsynteny analysis module but also greatly fills a crucial gap in facilitating genome macrosynteny analysis. Furthermore, it innovatively proposes the systematic integration of microsynteny, macrosynteny, and functional genomics data. For me, the entire process has been difficult and filled with both challenges and joy. I am also fortunate to have received Prof. Li's guidance on script writing and other learning skills.
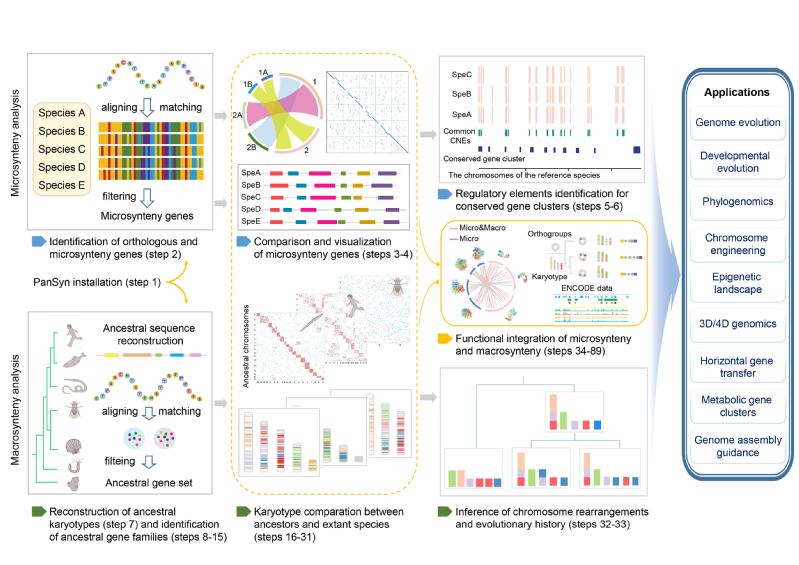
Figure 1: Schematic overview of PanSyn approach for comprehensive microsynteny and macrosynteny analysis.
We are excited to have developed PanSyn and eager to see how PanSyn will promote innovative discoveries and breakthroughs in the field of life sciences and depicts the evolution of the tree of life. In mid-December 2023, I had the honor of attending the 3rd AsiaEvo conference in Singapore. I was given the opportunity to make an oral presentation about PanSyn. After my presentation, I received significant interest and recognition from many attendees regarding our project. I am sincerely grateful to them for affirming the value of our work. Their support serves as a powerful driving force for me to conduct more impactful scientific research in the future.
When looking back, I am immensely grateful to Prof. Wang and Prof. Li for providing continuous guidance and instilling confidence in me throughout the entire project. Thanks also to all team members in the lab of Genetics & Evolution of Molluscs (http://www.molevolab.com) for their full support. Additionally, I would also like to express my deepest appreciation to the scientists who have dedicated their efforts to genome synteny research. Lastly, I am sincerely thankful to the numerous genome sequencing projects that have presented us with both challenges and opportunities.
We would love for people to give the protocol a try to study genome synteny in their favorite species or groups! For more information, you can find the paper in Nature Protocols, and the PanSyn pipeline is available on GitHub (https://github.com/yhw320/PanSyn).
References
1. Lemons, D. & McGinnis, W. Genomic evolution of Hox gene clusters. Science 313, 1918–1922 (2006).
2. Putnam, N. H. et al. The amphioxus genome and the evolution of the chordate karyotype. Nature 453, 1064–1071 (2008).
3. Wang, S. et al. Scallop genome provides insights into evolution of bilaterian karyotype and development. Nat. Ecol. Evol. 1, 120 (2017).
4. Simakov, O. et al. Deeply conserved synteny resolves early events in vertebrate evolution. Nat. Ecol. Evol. 4, 820–830 (2020).
5. Simion, P. et al. Chromosome-level genome assembly reveals homologous chromosomes and recombination in asexual rotifer Adineta vaga. Sci. Adv. 7, eabg4216 (2021).
6. Nguyen, N. T. T., Vincens, P., Dufayard, J. F., Roest Crollius, H. & Louis, A. Genomicus in 2022: comparative tools for thousands of genomes and reconstructed ancestors. Nucleic Acids Res. 50, D1025–D1031 (2022).
7. Bao, Y. et al. Genomic Insights into the Origin and Evolution of Molluscan Red-Bloodedness in the Blood Clam Tegillarca granosa. Mol Biol Evol. 38, 2351-2365 (2021).
8. Li, Y. et al. Adaptive bird-like genome miniaturization during the evolution of scallop swimming lifestyle. Genomics Proteomics Bioinformatics. 20, 1066–1077 (2022).
9. Han, W. et al. Ancient homomorphy of molluscan sex chromosomes sustained by reversible sex-biased genes and sex determiner translocation. Nat. Ecol. Evol. 6, 1891–1906 (2022).
10. Liu, F. et al. MolluscDB: an integrated functional and evolutionary genomics database for the hyper-diverse animal phylum Mollusca. Nucleic Acids Res. 49(D1), D988-D997 (2021).
11. Wei, J. et al. EDomics: a comprehensive and comparative multi-omics database for animal evo–devo. Nucleic Acids Res. 51, D913–D923 (2023).
12. Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49 (2012).
13. Haas, B. J. et al. DAGchainer: a tool for mining segmental genome duplications and synteny. Bioinformatics 20, 3643–3646 (2004).
14. Soderlund, C., Bomhoff, M. & Nelson, W.M. SyMAP v3.4: a turnkey synteny system with application to plant genomes. Nucleic Acids Res. 39, e68 (2011).
15. Simakov, O. et al. Deeply conserved synteny and the evolution of metazoan chromosomes. Sci. Adv. 8, eabi5884 (2022).
16. Xiao, Z. & Lam, H. M. ShinySyn: a Shiny/R application for the interactive visualization and integration of macro- and micro-synteny data. Bioinformatics 38, 4406–4408 (2022).
17. Robert, N. S. M., Sarigol, F., Zieger, E. & Simakov, O. SYNPHONI: scale-free and phylogeny-aware reconstruction of synteny conservation and transformation across animal genomes. Bioinformatics 38, 5434–5436 (2022).
18. Przybyla, L., Gilbert, L.A. A new era in functional genomics screens. Nat Rev Genet. 23, 89–103 (2022).

